泊洛沙姆188
Poluoshamu 188
Poloxamer 188
H(C2H4O)a(C3H6O)b(C2H4O)aOH
本品为α-氢-ω-羟基聚(氧乙烯)a-聚(氧丙烯)b-聚(氧乙烯)a嵌段共聚物。由环氧丙烷和丙二醇反应,形成聚氧丙烯二醇,然后加入环氧乙烷形成嵌段共聚物。在共聚物中氧乙烯单元(a)为75~85,氧丙烯单元(b)为25~30,氧乙烯(EO)含量为79.9%~83.7%,平均分子量为7680~9510。
【性状】本品为白色或类白色蜡状固体。
本品在水或乙醇中易溶,在无水乙醇或乙酸乙酯中溶解。
【鉴别】本品的红外光吸收图谱应与对照图谱(附图)一致(通则0402)。
【检查】酸碱度 取本品1.0g,加水10ml溶解后,依法测定(通则0631),pH值应为5.0~7.5。
溶液的澄清度与颜色 取酸碱度项下的溶液,依法检查(通则0901与通则0902),应澄清无色。
氧乙烯 取本品0.1~0.2g,用含1%4,4-二甲基-4-硅杂戊磺酸钠的氘代水1ml或者含四甲基硅烷的氘代三氯甲烷1ml溶解;将样品溶液装入核磁共振管中,如果是以含四甲基硅烷的氘代三氯甲烷为溶剂,加氘代水1滴,振摇,在核磁共振仪中,从0到5×10-6扫描,以直接比较法定量,按下式计算氧乙烯(EO)值:
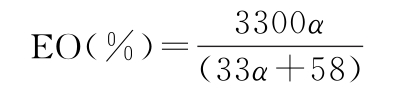
式中 α=(A2/A1)-1
A1为约1.15×10-6处峰的积分面积,代表氧丙烯的甲基;
A2为(3.2~3.8)×10-6处复合峰的积分面积,代表氧丙烯、氧乙烯的CH2O和氧丙烯的CHO;
EO为氧乙烯在整个分子组成中所占的比例,应为79.9%~83.7%。
不饱和度 称取研细后的本品约15.0g,精密加醋酸汞溶液50ml,在磁力搅拌下使完全溶解,静置30分钟,间断振摇,加溴化钠结晶10g,在磁力搅拌下混合2分钟,立即加酚酞指示液1ml,用甲醇制氢氧化钾滴定液(0.1mol/L)滴定,以空白试验和初始酸度校正[取泊洛沙姆15.0g,加中性甲醇(对酚酞指示液显中性)75ml溶解后,用甲醇制氢氧化钾滴定液(0.1mol/L)中和至对酚酞指示液显中性]。用下式计算不饱和度(mmol/g),不得过0.034mmol/g。

式中 V为供试品、空白和初始酸度消耗的甲醇制氢氧化钾滴定液(0.1mol/L)的体积,ml;
N为甲醇制氢氧化钾滴定液的浓度,mol/L;
W为供试品重量,g。
平均分子量 取本品适量(约相当于分子量×0.002g),精密称定,精密加邻苯二甲酸酐-吡啶溶液25ml,再加少许沸石,加热回流1小时,放冷,用吡啶冲洗冷凝器两次,每次10ml,加水10ml,混匀,加塞放置10分钟,精密加0.66mol/L氢氧化钠溶液50ml,再加酚酞-吡啶溶液(1→100)0.5ml,用氢氧化钠滴定液(0.5mol/L)滴定,显微粉红色,持续15秒不褪色,并将滴定的结果用空白试验校正,即得。按下式计算供试品的平均分子量,应为7680~9510。

式中 W为供试品重量,g;
B为空白消耗氢氧化钠滴定液(0.5mol/L)的体积,ml;
S为供试品消耗氢氧化钠滴定液(0.5mol/L)的体积,ml;
N为氢氧化钠滴定液的浓度,mol/L。
环氧乙烷、环氧丙烷与1,4-二氧六环 取本品1g,精密称定,置顶空瓶中,精密加水5ml,密封,作为供试品溶液。
另取环氧乙烷、环氧丙烷与1,4-二氧六环适量,用水溶解并定量稀释制成每1ml中含0.2μg、1μg与1μg的混合溶液,精密量取5ml,置顶空瓶中,密封,作为对照品溶液。
照气相色谱法(通则0521)测定,用6%氰丙基苯基-94%二甲基聚硅氧烷(或极性相近)为固定液的毛细管柱为色谱柱(0.25mm×30m, 1.40μm),起始温度为35℃,维持5分钟,以每分钟5℃的速率升温至120℃,维持5分钟,再以每分钟35℃的速率升温至220℃,维持5分钟;进样口温度为250℃;检测器温度为280℃;顶空瓶平衡温度为80℃,平衡时间为30分钟。取对照品溶液顶空进样,环氧乙烷峰、环氧丙烷峰与1,4-二氧六环峰之间的分离度应符合要求。再取供试品溶液与对照品溶液分别顶空进样,记录色谱图。按外标法以峰面积计算,含环氧乙烷不得过0.0001%,环氧丙烷不得过0.0005%,1,4-二氧六环不得过0.0005%。
乙二醇、二甘醇与三甘醇 取本品1.0g,精密称定,置10ml量瓶中,精密加内标溶液(取1,3-丁二醇适量,精密称定,加乙醇溶解并定量稀释制成每1ml中含0.1mg的溶液)1ml,用乙醇稀释至刻度,摇匀,作为供试品溶液。
另取乙二醇、二甘醇与三甘醇对照品适量,精密称定,用乙醇溶解并定量稀释制成每1ml中各含0.1mg的混合溶液,精密量取1ml,置10ml量瓶中,精密加内标溶液1ml,用乙醇稀释至刻度,摇匀,作为对照品溶液。
照气相色谱法(通则0521)测定,用50%苯基-50%二甲基聚硅氧烷(或极性相近)为固定液的毛细管柱为色谱柱(0.53mm×30m, 1.0μm),起始温度为60℃,维持5分钟,以每分钟10℃的速率升温至100℃,再以每分钟4℃的速率升温至170℃,最后以每分钟30℃的速率升温至290℃,维持30分钟;进样口温度为270℃;检测器温度为290℃。
精密量取供试品溶液和对照品溶液各1μl,分别注入气相色谱仪,记录色谱图。按内标法以峰面积计算,含乙二醇、二甘醇与三甘醇均不得过0.01%。
二丁基羟基甲苯(标示含二丁基羟基甲苯时测定) 取本品适量,精密称定,加乙醇溶解并定量稀释制成每1ml中含50mg的溶液,作为供试品溶液。
另取二丁基羟基甲苯对照品适量,精密称定,加乙醇溶解并定量稀释制成每1ml中含10μg的溶液,作为对照品溶液。
照气相色谱法(通则0521)测定,以50%苯基-50%二甲基聚硅氧烷(或极性相近)为固定液的毛细管柱为色谱柱(0.53mm×30m, 1.0μm),起始温度为60℃,维持5分钟,以每分钟10℃的速率升温至100℃,再以每分钟4℃的速率升温至180℃,最后以每分钟30℃的速率升温至290℃,维持30分钟;进样口温度为270℃;检测器温度为290℃。
精密量取供试品溶液与对照品溶液各2μl,分别注入气相色谱仪,记录色谱图。按外标法以峰面积计算,含二丁基羟基甲苯不得过0.02%。
水分 取本品,照水分测定法(通则0832第一法1)测定,含水分不得过1.0%。
炽灼残渣 取本品1.0g,依法检查(通则0841),遗留残渣不得过0.4%。
重金属 取炽灼残渣项下遗留的残渣,依法检查(通则0821第二法),含重金属不得过百万分之二十。
砷盐 取本品1.0g,加盐酸5ml与水23ml,振摇使溶解,依法检查(通则0822第一法),应符合规定(0.0002%)。
细菌内毒素(供注射用) 取本品,依法检查(通则1143),每1mg中含内毒素的量应小于0.012EU。
【类别】增溶剂和乳化剂等。
【贮藏】遮光,密闭保存。
【标示】如加抗氧剂,应标明抗氧剂名称与用量。