 药智官方微信
药智官方微信
 药智医械公众号
药智官方微信
药智医械公众号
药智医械公众号
药智官方微信
药智医械公众号

 该商家已通过实名认证
该商家已通过实名认证
巴西棕榈蜡
Baxi Zonglüla
Carnauba Wax
[8015-86-9]
本品系从Copernicia cerifera Mart.叶子中提取纯化而制得的蜡。
【性状】本品为淡黄色或黄色粉末、薄片或块状物。
本品在水或乙醇中几乎不溶。
熔点 本品的熔点(通则0612第二法)为80~86℃。
酸值 取本品约5g,精密称定,置250ml锥形瓶中,加二甲苯100ml,加热至完全溶解,加乙醇50ml和溴麝香草酚蓝指示液2.5ml,加热使澄清后,趁热用乙醇制氢氧化钾滴定液(0.1mol/L)滴至溶液显绿色,并将滴定结果用空白试验校正。酸值(通则0713)应为2~7。
碘值 取本品约1.8g,精密称定,置500ml干燥碘瓶中,加三氯甲烷30ml,在80℃±1℃水浴中加热溶解后,依法测定(通则0713),碘值应为5~14。
皂化值 取本品约3g,精密称定,置500ml锥形瓶中,加异丙醇-甲苯(5∶4)混合液50ml,精密加0.5mol/L氢氧化钾乙醇溶液15ml,加热回流3小时,加酚酞指示液1ml,趁热用盐酸滴定液(0.5mol/L)滴定,至溶液粉红色刚好褪去,加热至沸,如溶液又出现粉红色,再滴定至粉红色刚好褪去,并将滴定的结果用空白试验校正。皂化值(通则0713)应为78~95。
【鉴别】 取本品约0.1g,加三氯甲烷5ml,加热溶解,作为供试品溶液(趁热点样);另取薄荷醇、麝香草酚各约10mg与乙酸薄荷酯10μl,置同一20ml量瓶中,加甲苯稀释至刻度,摇匀,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液6μl与对照品溶液2μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-三氯甲烷(2∶98)为展开剂,展开,取出,晾干,喷以新制的20%磷钼酸乙醇溶液,在105℃加热10~15分钟至斑点清晰,立即检视。对照品溶液显示的斑点由低至高依次为深蓝色的薄荷醇、红色的麝香草酚和深蓝色的乙酸薄荷酯。供试品溶液应在薄荷醇与麝香草酚相应的位置之间显示一个大的斑点(三十烷烃),其下方可见多个微小斑点,在麝香草酚与乙酸薄荷酯相应的位置之间显示多个蓝色斑点,在上述斑点之上还应显示其他斑点,比移值(Rf)最大的斑点应清晰,原点应显蓝色。
【检查】溶液的澄清度与颜色 取本品0.1g,加三氯甲烷10ml,加热使溶解,依法检查(通则0901与通则0902),溶液应澄清无色;如显色,与同体积的对照液(取比色用重铬酸钾液1.0ml,加水15ml,摇匀,即得)比较,不得更深。
炽灼残渣 取本品1.0g,依法测定(通则0841),遗留残渣不得过0.25%。
重金属 取炽灼残渣项下遗留的残渣,依法检查(通则0821),含重金属不得过百万分之二十。
【类别】包衣剂和释放调节剂等。
【贮藏】密封保存。
巴西棕榈蜡

联系电话:18892005891
辛酸钠
Xinsuanna
Sodium Caprylate

C8H15NaO2 166.20
[1984-06-1]
本品按无水物计算,含C8H15NaO2不得少于99.0%。
【性状】本品为白色或类白色结晶性粉末。
本品在水或冰醋酸中易溶,在乙醇中略溶,在丙酮中几乎不溶。
【鉴别】(1)取本品约20mg,加水0.5ml溶解后,加甲氧基苯乙酸试液(取甲氧基苯乙酸2.7g,加10%氢氧化四甲铵的甲醇溶液6ml溶解后,加乙醇20ml,摇匀,贮存于聚乙烯容器中)1.5ml,于冰浴中冷却30分钟,生成大量白色结晶性沉淀;置20℃的水浴中,搅拌5分钟,沉淀不消失;加氨试液1ml,沉淀完全溶解;再加16%碳酸铵溶液1ml,没有沉淀生成。
(2)在有关物质项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
【检查】碱度 取本品2.5g,加水25ml溶解后,依法测定(通则0631),pH值应为8.0~10.5。
溶液的澄清度与颜色 取本品2.5g,加水25ml溶解后,依法检查(通则0901与通则0902),溶液应澄清无色;如显色,与橙黄色1号标准比色液(通则0901第一法)比较,不得更深。
水分 取本品,照水分测定法(通则0832第一法1)测定,含水分不得过3.0%。
重金属 取本品2.0g,加冰醋酸-水-乙醇(5∶10∶85)溶解并稀释至25ml,依法检查(通则0821第一法),含重金属不得过百万分之五。
有关物质 取本品约0.12g,加水5ml溶解后,加稀硫酸1ml,摇匀,加乙酸乙酯10ml,振摇提取后,静置使分层,取乙酸乙酯层,加无水硫酸钠干燥后,取上清液作为供试品溶液。
精密量取供试品溶液1ml,置100ml量瓶中,用乙酸乙酯稀释至刻度,摇匀,精密量取5ml,置50ml量瓶中,用乙酸乙酯稀释至刻度,摇匀,作为对照溶液。
另取辛酸对照品约10mg,精密称定,加乙酸乙酯10ml使溶解,作为对照品溶液。
照气相色谱法(通则0521)测定,以2-硝基对苯二酸改性的聚乙二醇20M(或极性相近)为固定液的毛细管柱为色谱柱(0.25mm×30m, 0.25μm或效能相当的色谱柱);起始温度为100℃,维持1分钟,以每分钟5℃的速率升温至220℃,维持20分钟;进样口温度为250℃;检测器温度为250℃。取对照溶液1μl注入气相色谱仪,主成分色谱峰的信噪比应不少于5。取供试品溶液1μl注入气相色谱仪,记录色谱图,按面积归一化法计算。供试品溶液色谱图中如有杂质峰,单个杂质不得过0.3%,总杂质不得过0.5%。供试品溶液色谱图中任何小于对照溶液中主峰面积0.5倍的峰忽略不计。
细菌内毒素(供注射用) 取本品,依法检查(通则1143),每1mg辛酸钠中含内毒素的量应小于0.3EU。
【含量测定】取本品约0.15g,精密称定,加冰醋酸50ml使溶解,照电位滴定法(通则0701),用高氯酸滴定液(0.1 mol/L)滴定,并将滴定的结果用空白试验校正。每1 ml高氯酸滴定液(0.1 mol/L)相当于1 6.62mg的C8H15NaO2。
【类别】稳定剂和抑菌剂等。
【贮藏】密闭,凉暗处保存。
联系电话:18892005891

单双硬脂酸甘油酯
Danshuang Yingzhisuan Ganyouzhi
Glyceryl Mono and Distearate
本品为单、二、三硬脂酸和棕榈酸混合甘油酯。由硬脂酸与过量甘油通过酯化反应制得,或由氢化植物油与甘油在催化剂的作用下,经过醇解反应制得。含单甘油酯应为40.0%~55.0%,二甘油酯应为30.0%~45.0%,三甘油酯应为5.0%~15.0%。
【性状】本品为白色或类白色的蜡状颗粒或薄片。
本品在水中几乎不溶。
熔点 本品的熔点(通则0612第二法)为54~66℃。
酸值 取本品4.0g,精密称定,加乙醇-乙醚(1∶1)混合液50ml,缓慢加热回流使溶解,依法测定(通则0713),酸值应不大于3.0。
碘值 取本品1.0g,精密称定,加三氯甲烷15ml,振摇使溶解,依法测定(通则0713),碘值应不大于3.0。
皂化值 取本品2.0g,精密称定,依法测定(通则0713),皂化值应为158~177。
【鉴别】(1)取本品和单双硬脂酸甘油酯对照品,分别加三氯甲烷制成每1ml中约含50mg的溶液,作为供试品溶液和对照品溶液。
照薄层色谱法(通则0502)试验,吸取上述两种溶液各10μl,分别点于同一硅胶G薄层板上,以正己烷-乙醚(30∶70)为展开剂,展开,晾干,喷以罗丹明B的乙醇溶液(1→10 000),置紫外光灯(365nm)下检视。
对照品溶液应显四个完全分离的清晰斑点,供试品溶液所显的斑点的位置与颜色应与对照品溶液的四个斑点一致。
(2)在脂肪酸组成项下记录的色谱图中,供试品溶液中棕榈酸甲酯峰、硬脂酸甲酯峰的保留时间应分别与对照品溶液中相应峰的保留时间一致。
【检查】游离甘油 取含量测定项下的供试品溶液作为供试品溶液。
另取甘油对照品适量,精密称定,加四氢呋喃溶解并分别定量稀释制成每1ml中约含0.5mg、1.0mg、2.0mg、4.0mg的溶液,作为系列标准曲线用溶液。
照含量测定项下的色谱条件,精密量取上述系列标准曲线用溶液各40μl,分别注入液相色谱仪,记录色谱图,以峰面积与相应浓度计算直线回归方程,相关系数(r)应不小于0.995。
精密量取供试品溶液40μl,注入液相色谱仪,记录色谱图,由直线回归方程计算供试品溶液中的甘油含量,含游离甘油不得过6.0%。
水分 取本品,研细,以三氯甲烷-无水甲醇(1∶1)为溶剂,照水分测定法(通则0832第一法1)测定,含水分不得过1.0%。
炽灼残渣 取本品1.0g,依法检查(通则0841),遗留残渣不得过0.1%。
重金属 取炽灼残渣项下遗留的残渣,依法检查(通则0821第二法),含重金属不得过百万分之十。
镍 对照品溶液的制备 精密量取镍标准溶液(1.000g/L)1ml,置200ml量瓶中,用水稀释至刻度,摇匀,精密量取5ml,置50ml量瓶中,用水稀释至刻度,摇匀,精密量取0.2ml、0.5ml、1ml、1.5ml与2ml,分别置50ml量瓶中,分别加硝酸12ml,用水稀释至刻度,摇匀,即得;另取硝酸12ml,用水稀释至50ml,摇匀,作为对照品空白溶液。
供试品溶液的制备 取本品约0.25g,精密称定,置消解罐中,加硝酸6ml与浓过氧化氢溶液(30%)2ml,置微波消解仪中进行消解处理,结束后取出消解罐,放冷,补加浓过氧化氢溶液(30%)1~2ml,再次进行微波消解,结束后取出消解罐,放冷,用水将内容物定量转移至25ml量瓶中,用水稀释至刻度,摇匀,即得;同法制备供试品空白溶液。
测定法 取对照品空白溶液、对照品溶液、供试品空白溶液与供试品溶液,以石墨炉为原子化器,照原子吸收分光光度法(通则0406第一法)在232.0nm波长处测定,含镍量不得过0.0001%。
脂肪酸组成 取本品0.1g,依法测定(通则0713);分别取棕榈酸甲酯、硬脂酸甲酯和油酸甲酯适量,加正庚烷溶解并稀释制成每1ml中各含0.1mg的溶液,作为对照品溶液。
按面积归一化法计算,含硬脂酸不得少于40.0%,棕榈酸和硬脂酸总量不得少于90.0%。
【含量测定】照分子排阻色谱法(通则0514)测定。
色谱条件与系统适用性试验 用苯乙烯-二乙烯基苯共聚物为填充剂(7.8mm×300mm, 5μm的两根色谱柱串联或效能相当的色谱柱);以四氢呋喃为流动相;示差折光检测器。三甘油酯、二甘油酯、单甘油酯与甘油依次出峰(参考色谱图见附图)。二甘油酯峰与单甘油酯峰的分离度应符合要求,二甘油酯峰与三甘油酯峰的分离度不得小于1.0。
测定法 取本品适量,精密称定,加流动相溶解并定量稀释制成每1ml中约含40mg的溶液(如浑浊,滤过后取续滤液),精密量取40μl注入液相色谱仪,记录色谱图(附图),按下列公式分别计算单甘油酯、二甘油酯与三甘油酯的含量。

式中 A为游离甘油项下测定结果,%;
B为水分项下测定结果,%;
C为游离脂肪酸计算结果,%;
X为单甘油酯和游离脂肪酸峰面积之和;
Y为二甘油酯峰面积;
Z为三甘油酯峰面积。
【类别】乳化剂和增稠剂等。
【贮藏】遮光,密封保存。

薄荷脑
Bohenao
L-Menthol
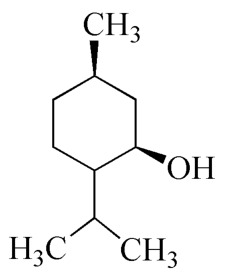
C10H20O 156.27
[1490-04-6]或[89-78-1]
本品为L-1-甲基-4-异丙基环己醇-3,系自唇形科植物薄荷Mentha haplocalyx Briq.的新鲜茎和叶经水蒸气蒸馏、冷冻、重结晶制得。含C10H20O应为95.0%~105.0%。
【性状】本品为无色针状或棱柱状结晶或白色结晶性粉末。
本品在乙醇中极易溶解,在水中极微溶解。
熔点 本品的熔点(通则0612)为42~44℃。
比旋度 取本品,精密称定,加乙醇溶解并定量稀释制成每1ml约含0.1g的溶液,依法测定(通则0621),比旋度为-49°至-50°。
【鉴别】(1)取本品1g,加硫酸20ml使溶解,即显橙红色,24小时后析出无薄荷脑香气的无色油层(与麝香草酚的区别)。
(2)取本品50mg,加冰醋酸1ml使溶解,加硫酸6滴与硝酸1滴的冷混合液,仅显淡黄色(与麝香草酚的区别)。
【检查】有关物质 取本品适量,精密称定,加无水乙醇溶解并定量稀释制成每1ml约含50mg的溶液,作为供试品溶液。
另取薄荷脑对照品适量,精密称定,加无水乙醇溶解并定量稀释制成每1ml约含0.5mg的溶液,作为对照品溶液。
照含量测定项下的色谱条件,其中柱温为110℃,取对照品溶液1μl注入气相色谱仪,记录色谱图,主成分峰高的信噪比应大于10;再精密量取供试品溶液与对照品溶液各1μl,分别注入气相色谱仪,记录色谱图至主成分峰保留时间的2倍。供试品色谱图中如有杂质峰,各杂质峰面积的和不得大于对照品溶液的主峰面积(1.0%)。
不挥发物 取本品2g,置已干燥至恒重的蒸发皿中,在水浴上加热,使缓缓挥散后,在105℃干燥至恒重,遗留残渣不得过1mg。
重金属与有害元素 照铅、镉、砷、汞、铜测定法(通则2321)测定,含铅不得过0.0005%,镉不得过0.000 03%,砷不得过0.0002%,汞不得过0.000 02%,铜不得过0.002%。
【含量测定】照气相色谱法(通则0521)测定。
色谱条件与系统适用性试验 用交联键合聚乙二醇(或极性相近)为固定液的毛细管柱为色谱柱;柱温为120℃;进样口温度为250℃;检测器温度为250℃。理论板数按薄荷脑峰计算不低于10 000。
测定法 取本品10mg,精密称定,置10ml量瓶中。加无水乙醇溶解并稀释至刻度,摇匀,作为供试品溶液,精密量取1μl注入气相色谱仪,记录色谱图;另取薄荷脑对照品,同法测定。按外标法以峰面积计算,即得。
【类别】矫味剂和芳香剂等。
【贮藏】密封,置阴凉处。
注:①本品有薄荷的特殊香气。②本品在乙醇溶液显中性反应。
单双硬脂酸甘油酯 Danshuang Yingzhisuan Ganyouzhi Glyceryl Mono and Distearate 本品为单、二、三硬脂酸和棕榈酸混合甘油酯。由硬脂酸与过量甘油通过酯化反应制得,或由氢化植物油与甘油在催化剂的作用下,经过醇解反应制得。含单甘油酯应为40.0%~55.0%,二甘油酯应为30.0%~45.0%,三甘油酯应为5.0%~15.0%。 【性状】本品为白色或类白色的蜡状颗粒或薄片。 本品在水中几乎不溶。 熔点 本品的熔点(通则0612第二法)为54~66℃。 酸值 取本品4.0g,精密称定,加乙醇-乙醚(1∶1)混合液50ml,缓慢加热回流使溶解,依法测定(通则0713),酸值应不大于3.0。 碘值 取本品1.0g,精密称定,加三氯甲烷15ml,振摇使溶解,依法测定(通则0713),碘值应不大于3.0。 皂化值 取本品2.0g,精密称定,依法测定(通则0713),皂化值应为158~177。 【鉴别】(1)取本品和单双硬脂酸甘油酯对照品,分别加三氯甲烷制成每1ml中约含50mg的溶液,作为供试品溶液和对照品溶液。 照薄层色谱法(通则0502)试验,吸取上述两种溶液各10μl,分别点于同一硅胶G薄层板上,以正己烷-乙醚(30∶70)为展开剂,展开,晾干,喷以罗丹明B的乙醇溶液(1→10 000),置紫外光灯(365nm)下检视。 对照品溶液应显四个完全分离的清晰斑点,供试品溶液所显的斑点的位置与颜色应与对照品溶液的四个斑点一致。 (2)在脂肪酸组成项下记录的色谱图中,供试品溶液中棕榈酸甲酯峰、硬脂酸甲酯峰的保留时间应分别与对照品溶液中相应峰的保留时间一致。 【检查】游离甘油 取含量测定项下的供试品溶液作为供试品溶液。 另取甘油对照品适量,精密称定,加四氢呋喃溶解并分别定量稀释制成每1ml中约含0.5mg、1.0mg、2.0mg、4.0mg的溶液,作为系列标准曲线用溶液。 照含量测定项下的色谱条件,精密量取上述系列标准曲线用溶液各40μl,分别注入液相色谱仪,记录色谱图,以峰面积与相应浓度计算直线回归方程,相关系数(r)应不小于0.995。 精密量取供试品溶液40μl,注入液相色谱仪,记录色谱图,由直线回归方程计算供试品溶液中的甘油含量,含游离甘油不得过6.0%。 水分 取本品,研细,以三氯甲烷-无水甲醇(1∶1)为溶剂,照水分测定法(通则0832第一法1)测定,含水分不得过1.0%。 炽灼残渣 取本品1.0g,依法检查(通则0841),遗留残渣不得过0.1%。 重金属 取炽灼残渣项下遗留的残渣,依法检查(通则0821第二法),含重金属不得过百万分之十。 镍 对照品溶液的制备 精密量取镍标准溶液(1.000g/L)1ml,置200ml量瓶中,用水稀释至刻度,摇匀,精密量取5ml,置50ml量瓶中,用水稀释至刻度,摇匀,精密量取0.2ml、0.5ml、1ml、1.5ml与2ml,分别置50ml量瓶中,分别加硝酸12ml,用水稀释至刻度,摇匀,即得;另取硝酸12ml,用水稀释至50ml,摇匀,作为对照品空白溶液。 供试品溶液的制备 取本品约0.25g,精密称定,置消解罐中,加硝酸6ml与浓过氧化氢溶液(30%)2ml,置微波消解仪中进行消解处理,结束后取出消解罐,放冷,补加浓过氧化氢溶液(30%)1~2ml,再次进行微波消解,结束后取出消解罐,放冷,用水将内容物定量转移至25ml量瓶中,用水稀释至刻度,摇匀,即得;同法制备供试品空白溶液。 测定法 取对照品空白溶液、对照品溶液、供试品空白溶液与供试品溶液,以石墨炉为原子化器,照原子吸收分光光度法(通则0406第一法)在232.0nm波长处测定,含镍量不得过0.0001%。 脂肪酸组成 取本品0.1g,依法测定(通则0713);分别取棕榈酸甲酯、硬脂酸甲酯和油酸甲酯适量,加正庚烷溶解并稀释制成每1ml中各含0.1mg的溶液,作为对照品溶液。 按面积归一化法计算,含硬脂酸不得少于40.0%,棕榈酸和硬脂酸总量不得少于90.0%。 【含量测定】照分子排阻色谱法(通则0514)测定。 色谱条件与系统适用性试验 用苯乙烯-二乙烯基苯共聚物为填充剂(7.8mm×300mm, 5μm的两根色谱柱串联或效能相当的色谱柱);以四氢呋喃为流动相;示差折光检测器。三甘油酯、二甘油酯、单甘油酯与甘油依次出峰(参考色谱图见附图)。二甘油酯峰与单甘油酯峰的分离度应符合要求,二甘油酯峰与三甘油酯峰的分离度不得小于1.0。 测定法 取本品适量,精密称定,加流动相溶解并定量稀释制成每1ml中约含40mg的溶液(如浑浊,滤过后取续滤液),精密量取40μl注入液相色谱仪,记录色谱图(附图),按下列公式分别计算单甘油酯、二甘油酯与三甘油酯的含量。 蒲公英 式中 A为游离甘油项下测定结果,%; B为水分项下测定结果,%; C为游离脂肪酸计算结果,%; X为单甘油酯和游离脂肪酸峰面积之和; Y为二甘油酯峰面积; Z为三甘油酯峰面积。 【类别】乳化剂和增稠剂等。 【贮藏】遮光,密封保存。

蔗糖
Zhetang
Sucrose
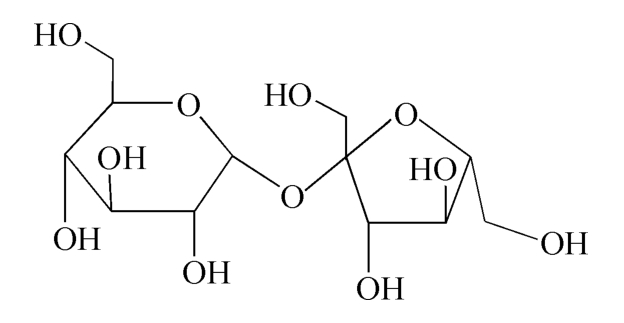
C12H22O11 342.30
[57-50-1]
本品为β-D-呋喃果糖基-α-D-吡喃葡萄糖苷。
【性状】本品为无色结晶或白色结晶性的松散粉末。
本品在水中极易溶解,在乙醇或无水乙醇中几乎不溶。
比旋度 取本品,精密称定,加水溶解并定量稀释制成每1ml中约含0.1g的溶液,依法测定(通则0621),比旋度为+66.3°至+67.0°。
【鉴别】(1)取本品,加0.05mol/L硫酸溶液,煮沸后,用0.1mol/L氢氧化钠溶液中和,再加碱性酒石酸铜试液,加热即生成氧化亚铜的红色沉淀。
(2)本品的红外光吸收图谱应与蔗糖对照品的图谱一致(通则0402)。
【检查】溶液的颜色 取本品5g,加水5ml溶解后,如显色,与黄色4号标准比色液(通则0901第一法)比较,不得更深。
硫酸盐 取本品1.0g,依法检查(通则0802),与标准硫酸钾溶液5.0ml制成的对照液比较,不得更浓(0.05%)。
还原糖 取本品5.0g,置250ml锥形瓶中,加水25ml溶解后,精密加碱性枸橼酸铜试液25ml与玻璃珠数粒,加热回流使在3分钟内沸腾,从全沸时起,连续沸腾5分钟,迅速冷却至室温(此时应注意勿使瓶中氧化亚铜与空气接触),立即加25%碘化钾溶液15ml,摇匀,随振摇随缓缓加入硫酸溶液(1→5)25ml,待二氧化碳停止放出后,立即用硫代硫酸钠滴定液(0.1mol/L)滴定,至近终点时,加淀粉指示液2ml,继续滴定至蓝色消失,同时做一空白试验。二者消耗硫代硫酸钠滴定液(0.1mol/L)的体积差不得过2.0ml(0.10%)。
炽灼残渣 取本品2.0g,依法检查(通则0841),遗留残渣不得过0.1%。
钙盐 取本品1.0g,加水25ml使溶解,加氨试液1ml与草酸铵试液5ml,摇匀,放置1小时,与钙标准溶液(精密称取碳酸钙0.125g,置500ml量瓶中,加水5ml与盐酸0.5ml使溶解,加水至刻度,摇匀。每1ml相当于0.10mg的Ca)5.0ml制成的对照液比较,不得更浓(0.05%)。
重金属 取炽灼残渣项下遗留的残渣,依法检查(通则0821第二法),含重金属不得过百万分之五。
【类别】矫味剂和黏合剂等。
【贮藏】密封,在干燥处保存。
巴西棕榈蜡
Baxi Zonglüla
Carnauba Wax
[8015-86-9]
本品系从Copernicia cerifera Mart.叶子中提取纯化而制得的蜡。
【性状】本品为淡黄色或黄色粉末、薄片或块状物。
本品在水或乙醇中几乎不溶。
熔点 本品的熔点(通则0612第二法)为80~86℃。
酸值 取本品约5g,精密称定,置250ml锥形瓶中,加二甲苯100ml,加热至完全溶解,加乙醇50ml和溴麝香草酚蓝指示液2.5ml,加热使澄清后,趁热用乙醇制氢氧化钾滴定液(0.1mol/L)滴至溶液显绿色,并将滴定结果用空白试验校正。酸值(通则0713)应为2~7。
碘值 取本品约1.8g,精密称定,置500ml干燥碘瓶中,加三氯甲烷30ml,在80℃±1℃水浴中加热溶解后,依法测定(通则0713),碘值应为5~14。
皂化值 取本品约3g,精密称定,置500ml锥形瓶中,加异丙醇-甲苯(5∶4)混合液50ml,精密加0.5mol/L氢氧化钾乙醇溶液15ml,加热回流3小时,加酚酞指示液1ml,趁热用盐酸滴定液(0.5mol/L)滴定,至溶液粉红色刚好褪去,加热至沸,如溶液又出现粉红色,再滴定至粉红色刚好褪去,并将滴定的结果用空白试验校正。皂化值(通则0713)应为78~95。
【鉴别】 取本品约0.1g,加三氯甲烷5ml,加热溶解,作为供试品溶液(趁热点样);另取薄荷醇、麝香草酚各约10mg与乙酸薄荷酯10μl,置同一20ml量瓶中,加甲苯稀释至刻度,摇匀,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液6μl与对照品溶液2μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-三氯甲烷(2∶98)为展开剂,展开,取出,晾干,喷以新制的20%磷钼酸乙醇溶液,在105℃加热10~15分钟至斑点清晰,立即检视。对照品溶液显示的斑点由低至高依次为深蓝色的薄荷醇、红色的麝香草酚和深蓝色的乙酸薄荷酯。供试品溶液应在薄荷醇与麝香草酚相应的位置之间显示一个大的斑点(三十烷烃),其下方可见多个微小斑点,在麝香草酚与乙酸薄荷酯相应的位置之间显示多个蓝色斑点,在上述斑点之上还应显示其他斑点,比移值(Rf)最大的斑点应清晰,原点应显蓝色。
【检查】溶液的澄清度与颜色 取本品0.1g,加三氯甲烷10ml,加热使溶解,依法检查(通则0901与通则0902),溶液应澄清无色;如显色,与同体积的对照液(取比色用重铬酸钾液1.0ml,加水15ml,摇匀,即得)比较,不得更深。
炽灼残渣 取本品1.0g,依法测定(通则0841),遗留残渣不得过0.25%。
重金属 取炽灼残渣项下遗留的残渣,依法检查(通则0821),含重金属不得过百万分之二十。
【类别】包衣剂和释放调节剂等。
【贮藏】密封保存。
巴西棕榈蜡

聚丙烯酸树脂Ⅱ
Jubingxisuan Shuzhi Ⅱ
Polyacrylic ResinⅡ
本品为甲基丙烯酸与甲基丙烯酸甲酯以1∶1的比例共聚而得。
【性状】本品为白色条状物或粉末,在乙醇中易结块。
本品(如为条状物断成长约1cm,粉末则不经研磨)在水中不溶。
酸值 取本品约0.5g,精密称定,置250ml锥形瓶中,加75%中性乙醇(对酚酞指示液显中性)25ml,微温使溶解,放冷,精密滴加氢氧化钠滴定液(0.1mol/L)15ml,加氯化钠5g与水10ml,用氢氧化钠滴定液(0.1mol/L)继续滴定至粉红色持续30秒不褪。本品的酸值(通则0713),按干燥品计算,应为300~330。
【鉴别】本品的红外光吸收图谱应与对照品的图谱一致(通则0402)。
【检查】黏度 取本品6.0g,加乙醇100ml,微温使溶解,用旋转黏度计,依法测定(通则0633第三法),在25℃时的动力黏度不得过50mPa·s。
酸度 取本品3.0g,加pH值约为7的75%乙醇100ml,微温使溶解,放冷,依法测定(通则0631),pH值应为4.0~6.0。
残留单体 照高效液相色谱法(通则0512)测定。
色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂,以甲醇-水(用磷酸调节pH值至2.0)(20∶80)为流动相,检测波长为202nm。甲基丙烯酸峰、甲基丙烯酸甲酯峰与相邻杂质峰间的分离度应符合要求。
磷酸盐缓冲液 配制含有1.78%无水磷酸氢二钠和1.7%磷酸二氢钾的溶液,用磷酸调节pH值至2.0。
对照品溶液 取甲基丙烯酸与甲基丙烯酸甲酯对照品各约0.05g,精密称定,加甲醇稀释制成每1ml中各约含5μg的混合溶液,精密量取3ml,置10ml量瓶中,用上述磷酸盐缓冲液稀释至刻度,摇匀,即得对照品溶液。
供试品溶液 取本品约0.5g,精密称定,加甲醇溶解并稀释至50ml,摇匀,精密量取3ml,置10ml量瓶中用磷酸盐缓冲液稀释至刻度,以每分钟12 000转的转速离心10分钟,取上清液滤过,取续滤液作为供试品溶液。
测定法 分别精密量取供试品溶液与对照品溶液各20μl,注入液相色谱仪,按外标法以峰面积计算,甲基丙烯酸与甲基丙烯酸甲酯的含量之和不得过0.1%。
干燥失重 取本品,在110℃干燥至恒重,减失重量不得过5.0%(通则0831)。
重金属 取本品1.0g,依法检查(通则0821第二法),含重金属不得过百万分之三十。
【类别】包衣剂和释放调节剂等。
【贮藏】密封,在30℃以下保存。

泊洛沙姆188
Poluoshamu 188
Poloxamer 188
H(C2H4O)a(C3H6O)b(C2H4O)aOH
本品为α-氢-ω-羟基聚(氧乙烯)a-聚(氧丙烯)b-聚(氧乙烯)a嵌段共聚物。由环氧丙烷和丙二醇反应,形成聚氧丙烯二醇,然后加入环氧乙烷形成嵌段共聚物。在共聚物中氧乙烯单元(a)为75~85,氧丙烯单元(b)为25~30,氧乙烯(EO)含量为79.9%~83.7%,平均分子量为7680~9510。
【性状】本品为白色或类白色蜡状固体。
本品在水或乙醇中易溶,在无水乙醇或乙酸乙酯中溶解。
【鉴别】本品的红外光吸收图谱应与对照图谱(附图)一致(通则0402)。
【检查】酸碱度 取本品1.0g,加水10ml溶解后,依法测定(通则0631),pH值应为5.0~7.5。
溶液的澄清度与颜色 取酸碱度项下的溶液,依法检查(通则0901与通则0902),应澄清无色。
氧乙烯 取本品0.1~0.2g,用含1%4,4-二甲基-4-硅杂戊磺酸钠的氘代水1ml或者含四甲基硅烷的氘代三氯甲烷1ml溶解;将样品溶液装入核磁共振管中,如果是以含四甲基硅烷的氘代三氯甲烷为溶剂,加氘代水1滴,振摇,在核磁共振仪中,从0到5×10-6扫描,以直接比较法定量,按下式计算氧乙烯(EO)值:
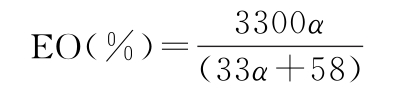
式中 α=(A2/A1)-1
A1为约1.15×10-6处峰的积分面积,代表氧丙烯的甲基;
A2为(3.2~3.8)×10-6处复合峰的积分面积,代表氧丙烯、氧乙烯的CH2O和氧丙烯的CHO;
EO为氧乙烯在整个分子组成中所占的比例,应为79.9%~83.7%。
不饱和度 称取研细后的本品约15.0g,精密加醋酸汞溶液50ml,在磁力搅拌下使完全溶解,静置30分钟,间断振摇,加溴化钠结晶10g,在磁力搅拌下混合2分钟,立即加酚酞指示液1ml,用甲醇制氢氧化钾滴定液(0.1mol/L)滴定,以空白试验和初始酸度校正[取泊洛沙姆15.0g,加中性甲醇(对酚酞指示液显中性)75ml溶解后,用甲醇制氢氧化钾滴定液(0.1mol/L)中和至对酚酞指示液显中性]。用下式计算不饱和度(mmol/g),不得过0.034mmol/g。

式中 V为供试品、空白和初始酸度消耗的甲醇制氢氧化钾滴定液(0.1mol/L)的体积,ml;
N为甲醇制氢氧化钾滴定液的浓度,mol/L;
W为供试品重量,g。
平均分子量 取本品适量(约相当于分子量×0.002g),精密称定,精密加邻苯二甲酸酐-吡啶溶液25ml,再加少许沸石,加热回流1小时,放冷,用吡啶冲洗冷凝器两次,每次10ml,加水10ml,混匀,加塞放置10分钟,精密加0.66mol/L氢氧化钠溶液50ml,再加酚酞-吡啶溶液(1→100)0.5ml,用氢氧化钠滴定液(0.5mol/L)滴定,显微粉红色,持续15秒不褪色,并将滴定的结果用空白试验校正,即得。按下式计算供试品的平均分子量,应为7680~9510。

式中 W为供试品重量,g;
B为空白消耗氢氧化钠滴定液(0.5mol/L)的体积,ml;
S为供试品消耗氢氧化钠滴定液(0.5mol/L)的体积,ml;
N为氢氧化钠滴定液的浓度,mol/L。
环氧乙烷、环氧丙烷与1,4-二氧六环 取本品1g,精密称定,置顶空瓶中,精密加水5ml,密封,作为供试品溶液。
另取环氧乙烷、环氧丙烷与1,4-二氧六环适量,用水溶解并定量稀释制成每1ml中含0.2μg、1μg与1μg的混合溶液,精密量取5ml,置顶空瓶中,密封,作为对照品溶液。
照气相色谱法(通则0521)测定,用6%氰丙基苯基-94%二甲基聚硅氧烷(或极性相近)为固定液的毛细管柱为色谱柱(0.25mm×30m, 1.40μm),起始温度为35℃,维持5分钟,以每分钟5℃的速率升温至120℃,维持5分钟,再以每分钟35℃的速率升温至220℃,维持5分钟;进样口温度为250℃;检测器温度为280℃;顶空瓶平衡温度为80℃,平衡时间为30分钟。取对照品溶液顶空进样,环氧乙烷峰、环氧丙烷峰与1,4-二氧六环峰之间的分离度应符合要求。再取供试品溶液与对照品溶液分别顶空进样,记录色谱图。按外标法以峰面积计算,含环氧乙烷不得过0.0001%,环氧丙烷不得过0.0005%,1,4-二氧六环不得过0.0005%。
乙二醇、二甘醇与三甘醇 取本品1.0g,精密称定,置10ml量瓶中,精密加内标溶液(取1,3-丁二醇适量,精密称定,加乙醇溶解并定量稀释制成每1ml中含0.1mg的溶液)1ml,用乙醇稀释至刻度,摇匀,作为供试品溶液。
另取乙二醇、二甘醇与三甘醇对照品适量,精密称定,用乙醇溶解并定量稀释制成每1ml中各含0.1mg的混合溶液,精密量取1ml,置10ml量瓶中,精密加内标溶液1ml,用乙醇稀释至刻度,摇匀,作为对照品溶液。
照气相色谱法(通则0521)测定,用50%苯基-50%二甲基聚硅氧烷(或极性相近)为固定液的毛细管柱为色谱柱(0.53mm×30m, 1.0μm),起始温度为60℃,维持5分钟,以每分钟10℃的速率升温至100℃,再以每分钟4℃的速率升温至170℃,最后以每分钟30℃的速率升温至290℃,维持30分钟;进样口温度为270℃;检测器温度为290℃。
精密量取供试品溶液和对照品溶液各1μl,分别注入气相色谱仪,记录色谱图。按内标法以峰面积计算,含乙二醇、二甘醇与三甘醇均不得过0.01%。
二丁基羟基甲苯(标示含二丁基羟基甲苯时测定) 取本品适量,精密称定,加乙醇溶解并定量稀释制成每1ml中含50mg的溶液,作为供试品溶液。
另取二丁基羟基甲苯对照品适量,精密称定,加乙醇溶解并定量稀释制成每1ml中含10μg的溶液,作为对照品溶液。
照气相色谱法(通则0521)测定,以50%苯基-50%二甲基聚硅氧烷(或极性相近)为固定液的毛细管柱为色谱柱(0.53mm×30m, 1.0μm),起始温度为60℃,维持5分钟,以每分钟10℃的速率升温至100℃,再以每分钟4℃的速率升温至180℃,最后以每分钟30℃的速率升温至290℃,维持30分钟;进样口温度为270℃;检测器温度为290℃。
精密量取供试品溶液与对照品溶液各2μl,分别注入气相色谱仪,记录色谱图。按外标法以峰面积计算,含二丁基羟基甲苯不得过0.02%。
水分 取本品,照水分测定法(通则0832第一法1)测定,含水分不得过1.0%。
炽灼残渣 取本品1.0g,依法检查(通则0841),遗留残渣不得过0.4%。
重金属 取炽灼残渣项下遗留的残渣,依法检查(通则0821第二法),含重金属不得过百万分之二十。
砷盐 取本品1.0g,加盐酸5ml与水23ml,振摇使溶解,依法检查(通则0822第一法),应符合规定(0.0002%)。
细菌内毒素(供注射用) 取本品,依法检查(通则1143),每1mg中含内毒素的量应小于0.012EU。
【类别】增溶剂和乳化剂等。
【贮藏】遮光,密闭保存。
【标示】如加抗氧剂,应标明抗氧剂名称与用量。

苯甲酸苄酯
Benjiasuanbianzhi
Benzyl Benzoate

C14H12O2 212.25
[120-51-4]
本品为苯甲酸苯甲酯。按无水物计算,含C14H12O2应为98.0%~102.0%。
【性状】本品为无色或几乎无色的油状液体。
相对密度 本品的相对密度(通则0601)在25℃时为1.116~1.120。
折光率 本品的折光率(通则0622)为1.568~1.570。
凝点 取本品15ml置内管中,加入少量母晶,使迅速冷却,并测定近似凝点。再将内管置约高于近似凝点10℃的水浴中,使凝结物仅剩极微量未熔融,依法测定(通则0613),在烧杯中加入约低于供试品近似凝点5℃的水或其他适宜的冷却液,用搅拌器不断搅拌,每隔30秒观察温度1次,至液体温度下降至恒定或开始上升时停止搅拌,并每隔5~10秒观察温度1次,至温度计汞柱在一点停留约1分钟不变,或微上升至最高温度后停留约1分钟不变,记录温度。连续读数次数应不少于4次,且各次读数相差应小于0.2℃,将平均值作为供试品的凝点,应不低于18.0℃。
【鉴别】(1)在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
(2)取本品,加乙醇溶解并稀释制成每1ml中约含5μg的溶液,照紫外-可见分光光度法(通则0401)测定,在230nm波长处有最大吸收。
(3)本品的红外光吸收图谱应与对照品的图谱一致(通则0402)。
【检查】酸度 取本品5.0g,置锥形瓶中,加中性乙醇25ml使溶解,摇匀,用氢氧化钠滴定液(0.02mol/L)滴定至显粉红色,消耗氢氧化钠滴定液(0.02mol/L)的体积不得过1.5ml。
氯化物 取本品2.0g,加水50ml, 80℃水浴加热5分钟,放冷,滤过;取续滤液5.0ml,依法检查(通则0801),与标准氯化钠溶液7.0ml制成的对照溶液比较,不得更浓(0.035%)。
有关物质 取本品适量,精密称定,加流动相溶解并定量稀释制成每1ml中约含1mg的溶液,作为供试品溶液。
精密量取供试品溶液1ml,置100ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。
取苯甲醛对照品适量,精密称定,加流动相溶解并定量稀释制成每1ml中约含0.5μg的溶液,作为对照品溶液。
称取苯甲醇与苯甲醛对照品各适量,加流动相溶解并稀释制成每1ml中约含苯甲醇1μg、苯甲醛0.5μg的混合溶液,作为系统适用性溶液。
精密量取对照溶液1ml,置50ml量瓶中,用流动相稀释至刻度,摇匀,作为灵敏度溶液。
照含量测定项下色谱条件,精密量取系统适用性溶液、灵敏度溶液、供试品溶液、对照品溶液与对照溶液各20μl,分别注入液相色谱仪,供试品溶液记录色谱图至主峰保留时间的两倍。系统适用性溶液色谱图中,两个主峰间的分离度应符合要求,理论板数按苯甲醛峰计算不低于3000。灵敏度溶液色谱图中,主成分峰的信噪比应大于10。
供试品溶液色谱图中如有与苯甲醛峰保留时间一致的色谱峰,按外标法以峰面积计算,不得过0.05%;如有其他杂质,不得大于对照溶液主峰面积的0.1倍(0.1%),其他各杂质峰面积的和不得大于对照溶液主峰面积(1.0%),小于灵敏度溶液主峰面积的峰忽略不计。
水分 取本品2.0g,照水分测定法(通则0832第一法1)测定,含水分不得过0.3%。
炽灼残渣 取本品1.0g,依法检查(通则0841),遗留残渣不得过0.05%。
细菌内毒素(供注射用) 取本品,加内毒素检查用水稀释至所需浓度,涡旋混合3分钟,离心,取水层,依法检查(通则1143),每1g苯甲酸苄酯中含内毒素的量应小于标示值。
【含量测定】照高效液相色谱法(通则0512)测定。
色谱条件与系统适用性试验 用苯基硅烷键合硅胶为填充剂,以乙腈-水(60∶40)为流动相,检测波长为254nm。
测定法 取本品适量,精密称定,加流动相溶解并定量稀释制成每1ml中约含0.2mg的溶液,作为供试品溶液,精密量取20μl注入液相色谱仪,记录色谱图;另取苯甲酸苄酯对照品适量,同法测定。按外标法以峰面积计算,即得。
【类别】增溶剂和溶剂等。
【贮藏】遮光,密闭保存。
苯甲酸苄酯
康洲大数据 Copyright © 2009-2026 药智网YAOZH.COM All Rights Reserved. 工信部备案号:渝ICP备10200070号-3
客户服务热线:400-678-0778 投诉建议:023-62988285转8001 E-mail:admin@yaozh.com
互联网增值电信业务许可证编号:渝B2-20120028 互联网药品信息服务资格证:(渝)-经营性-2021-0017

渝公网安备 50010802001068号

投诉热线: (023) 6262 8397
邮箱: tousu@yaozh.com
QQ: 236960938

