 药智官方微信
药智官方微信
 药智医械公众号
药智官方微信
药智医械公众号
药智医械公众号
药智官方微信
药智医械公众号




医疗器械定义及监管
1.医疗器械定义
医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的
计算机软件;其效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起辅助
作用,其目的是:
(一)疾病的诊断、预防、监护、治疗或者缓解;
(二)损伤的诊断、监护、治疗、缓解或者功能补偿;
(三)生理结构或者生理过程的检验、替代、调节或者支持;
(四)生命的支持或者维持;
(五)妊娠控制;
(六)通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。
2.医疗器械监管部门
国家药品监督管理局主管全国医疗器械注册与备案管理工作,负责建立医疗器械注册与备案管理工作体系和制度,依法组织境内第三类
和进口第二类、
第三类医疗器械审评审批,进口第一类医疗器械备案以及相关监督管理工作,对地方医疗器械注册与备案工作进行监督指导。
国家药品监督管理局医疗器械技术审评中心(以下简称国家局器械审评中心)负责需进行临床试验审批的医疗器械临床试验申请以及境内
第三类和进口
第二类、第三类医疗器械产品注册申请、变更注册申请、延续注册申请等的技术审评工作。
国家药品监督管理局医疗器械标准管理中心、中国食品药品检定研究院、国家药品监督管理局食品药品审核查验中心
(以下简称国家局审核查验中心),国家药品监督管理局药品评价中心、国家药品监督管理局行政事项受理服务和投诉举报中心、
国家药品监督管理局信息中心等其他专业技术机构,依职责承担实施医疗器械监督管理所需的医疗器械标准管理、分类界定、
检验、核查、监测与评价、制证送达以及相应的信息化建设与管理等相关工作。
省、自治区、直辖市药品监督管理部门负责本行政区域内以下医疗器械注册相关管理工作:
(一)境内第二类医疗器械注册审评审批;
(二)境内第二类、第三类医疗器械质量管理体系核查;
(三)依法组织医疗器械临床试验机构以及临床试验的监督管理;
(四)对设区的市级负责药品监督管理的部门境内第一类医疗器械备案的监督指导。
省、自治区、直辖市药品监督管理部门设置或者指定的医疗器械专业技术机构,承担实施医疗器械监督管理所需的技术审评、检验、核查监测与评价等工作。
设区的市级负责药品监督管理的部门负责境内第一类医疗器械产品备案管理工作。
医疗器械法规体系
1.医疗器械监管行政法规
我国在2000年1月颁布《医疗器械监督管理条例》,标志着医疗器械监管初步迈入法制化、规范化时代,其后在2014年和2020年先后
经过两次重要修订,内容体系逐步完善,立法质量不断提升,是目前医疗器械行业监管法规体系的核心,对医疗器械监管各方面的问题
做出了基本规定。
2023年9月,十四届全国人大常委会立法规划发布,《医疗器械管理法》首次被列入立法规划。
《医疗器械监督管理条例》属于行政法规,其效力层级低于法律,制约了监管制度的设计和实施。从立法的现实基础和国际经验方面看
《医疗器械管理法》是健全我国医药领域法律体系,补齐医疗器械监管短板的必然选择。
2.医疗器械监管部门规章
部门规章是指国务院相关部委在自己的职权范围内制订的法规。医疗器械的部门规章主要包括一下内容:

这些法规对医疗器械的研制、分类、临床试验、注册、生产、经营、使用、不良事件检查和再评价做出了针对性规定,是对《条例》内容的细化,
构成了医疗器械监管法规体系的主体。
3.医疗器械监管规范性文件
医疗器械规范性文件,是指部门规章外,医疗器械监管部门在法定职权内依法制订并公开发行的针对医疗器械的公告、通告、通知,
这类法规数量多、内容丰富、形式多样,是医疗器械行政法规和部门规章的重要补充,比如《关于发布第一类医疗器械产品目录的通告》《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》、《关于发布免于临床评价医疗器械目录的通告》等。
医疗器械行政法规、部门规章、规范性文件构成了医疗器械监督管理法规体系,其中行政法规是核心,部门规章是主题,
规范性文件是重要补充。
医疗器械分类
根据《医疗器械监督管理条例》规定,国家对医疗器械按照风险程度实行分类管理。
根据医疗器械的预期目的、结构特征、使用方法等因素,评价医疗器械风险程度,根据风险程度分为三类。
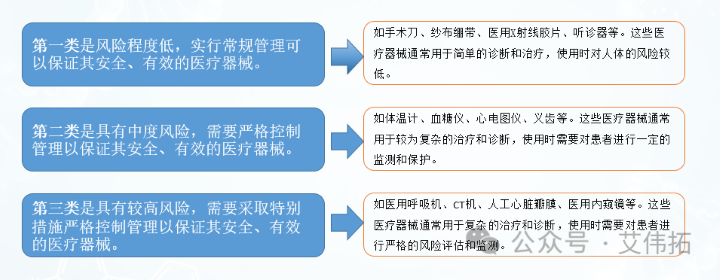
国务院药品监督管理部门负责制定医疗器械的分类规则和分类目录,并根据医疗器械生产、经营、使用情况,及时对医疗器械的风险化
进行分析、评价,对分类规则和分类目录进行调整.
我国医疗器械(含体外诊断试剂)分类实行分类规则指导下的分类目录制,分类规则和分类目录并存,以分类目录优先。
依据《医疗器械分类规则》及《体外诊断试剂分类规则》监管部门先后发布了《医疗器械分类目录》、《第一类医疗器械产品目录》,
以及《体外诊断试剂分类目录(修订草案征求意见稿)》,夯实医疗器械分类管理基础。
根据结构特征不同分类如下:

根据是否接触人体分类:

根据不同的结构特征和是否接触人体,医疗器械的使用形式包括:

根据不同的结构特征、是否接触人体以及使用形式,医疗器械的使用状态或者其产生的影响包括以下情形:
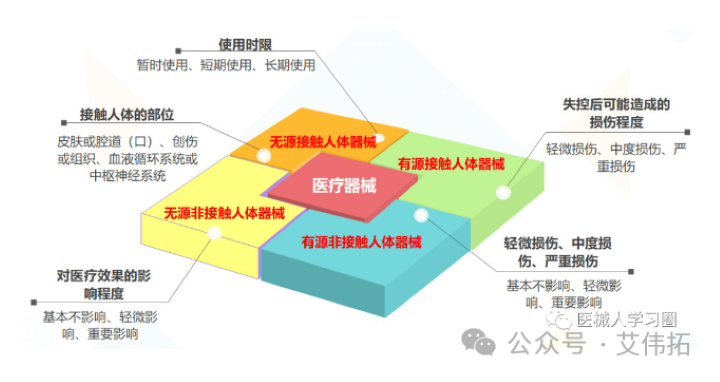
医疗器械分类的界定方法
医疗器械分类目录按照医疗器械技术专业和临床使用特点分为22个子目录,子目录由一级产品类别、二级产品类别、产品描述、预期
用途, 品名举例和管理类别组成。
判定产品类别时,应当根据产品的实际情况,结合新《分类目录》中产品描述、预期用途和品名举例进行综合判定。
医疗器械分类查询可以通过NMPA官网医疗器械栏-医疗器械查询,网站如下:

医疗器械分类查询也可以通过中检院官网-信息公开-数据查询-医疗器械分类目录查询。
根据《总局办公厅关于规范医疗器械产品分类有关工作的通知》,新研制的尚未列入《分类目录》或分类界定通知等文件的医疗器械,
按照《医疗器械监督管理条例》第十六条规定申请类别确认的,申请人应当通过总局医疗器械标准管理中心(以下简称标管中心)
分类界定信息系统提出分类界定申请。
申请材料要求如下:
(一)分类界定申请表;
(二)产品照片或产品结构图;
(三)产品技术要求及产品说明书(样稿);
(四)进口上市证明材料(如有);
(五)资料真实性自我保证声明;
(六)其他与产品分类界定有关的材料。
康洲大数据 Copyright © 2009-2026 药智网YAOZH.COM All Rights Reserved. 工信部备案号:渝ICP备10200070号-3
客户服务热线:400-678-0778 投诉建议:023-62988285转8001 E-mail:admin@yaozh.com
互联网增值电信业务许可证编号:渝B2-20120028 互联网药品信息服务资格证:(渝)-经营性-2021-0017

渝公网安备 50010802001068号

投诉热线: (023) 6262 8397
邮箱: tousu@yaozh.com
QQ: 236960938


